Unintended and Unattended Consequences: The Opportunity Costs of Reducing Exclusivity Rights for Intellectual Property
Published By: Matthias Bauer
Subjects: EU Single Market European Union Healthcare
Summary
The European Commission is reviewing the use of supplementary protection certificates (SPCs), a patent term extension motivated by the increasing length of market approvals for pharmaceuticals. The initiative is driven by EU Health Ministers thinking that changes in patent exclusivity rights will lower their expenditures on medicines. The generics industry’s push for a waiver has been backed by a promise to create additional jobs in the EU, but the evidence behind that promise is doubtful.
Available analyses suffer from a profound lack of appropriate data and disregard the opportunity costs of an SPC export waiver for EU Member States. In this paper, we will highlight that an SPC export waiver could prompt the EU’s innovative pharmaceutical companies to 1) reconsider their research and manufacturing activities in some EU Member States and 2) adapt product placement and pricing strategies in several high-income EU Member States to compensate for (the risk of) lower revenues. Given corporate strategies to overcome regulatory fragmentation, and experiences from market launch sequencing in the EU, an SPC export waiver could entail higher drug prices for governments and patients in Germany, the UK, France, the Nordics, and the Benelux.
The effective erosion of exclusivity rights in the EU would also come at the risk of losing innovative capacity and ‘high value-added jobs’ to other jurisdictions that offer a more attractive mix of IP protection and research and production costs. Moreover, the EU’s trade and investment policy has been to get other countries to allow patent term extension to compensate for revenue shortfalls due to longer regulatory approval times. If the EU allows for exports to these third markets when exclusivity rights are still active, it would erode the EU position vis-à-vis countries with weak protection of intellectual property.
Research assistance by Valentin Moreau is gratefully acknowledged.
Introduction
There is yet again a debate in Europe about the protection of intellectual property (IP) in the pharmaceutical sector, and it is influenced by the calls for introducing an export exemption for Supplementary Protection Certificates (SPCs). This paper aims to take stock of the arguments and evidence for and against such a reform, and to map the different opportunity costs that would entail from reduced exclusivity rights. While most other existing studies have come to the conclusion that static economic activity in the EU would only marginally be affected by an export waiver in the SPC system, this study highlights the dynamic effects that would arise. In essence, additional erosion in the exclusivity rights would disincentivize originators and innovators in Europe. Moreover, an SPC export waiver is likely to cause divestment and higher prices for branded medicines in some EU Member States, primarily more affluent Western European countries.
While the SPC is a technical and, for many observers, somewhat opaque system, it rests on a simple observation. Patents grant pharmaceutical manufacturers (patent holders) the temporary right to exclude others from making, using, or selling these drugs. The exclusive aim is to encourage innovation as patent holders are likely to collect greater revenues if they are protected from imitation and direct competition. These revenues are intended to serve as incentives for new research in substances and the development of innovative pharmaceutical products that could improve public health. However, the effective exclusivity right gradually eroded over time because of the compulsory, and increasingly lengthy, testing and clinical trials these products require prior to obtaining regulatory marketing approval. Hence, the economic value of the patent has gone down (see discussion below). An SPC is intended to address the negative impact on sales and revenues.
Over the years, regulators have gradually introduced more restrictions on drug developers in order to safeguard public health. Extensive regulatory systems have been constructed by bodies such as the European Medicines Agency (EMA) and the US Food and Drugs Administration (USFDA), prescribing procedures for virtually all stages along the drug development process (Kashyap et al. 2013; Putzeist 2013). Additional safety and pharmacovigilance requirements, for instance, were added partly as a consequence of previous market withdrawals. Minimum requirements for functional pharmacovigilance systems were set during a WHO-mediated process in 2010 and then implemented by regulators around the world (WHO 2012). In the EU, the new pharmacovigilance legislation, which entered into force in 2012, was the biggest change to the regulation of human medicines since 1995. The new regulation had significant implications for applicants and holders of EU marketing authorisations, as well as for patients, healthcare professionals and regulators (see, e.g., EMA 2012).
Mandatory trial and clinical testing times as well as the costs associated with these procedures increased substantially over time. Longer approval times and higher costs contributed to a significant decline in pharmaceutical research and development productivity for many therapeutic areas. Accordingly, the purpose of introducing SPC’s was to effectively stabilise the commercial value of a patent by granting pharmaceutical innovators additional time to recoup the costs for investments in the research and development of new medicines (for discussions of recent trends in regulatory affairs and marketing approval procedures see, e.g., Eichler et al. 2016; DiMasi 2016; Mestre-Ferrandiz et al. 2012; Scannell et al. 2012). As a consequence, the commercial value of a patent would be significantly lower if marketing exclusivity was not extended.
In the EU, SPCs must still be applied for and are granted at the Member State level. Once granted, the SPC takes effect at the end of the term of the original patent and extends exclusivity rights. The additional exclusivity period is equal to the period which elapsed between the date on which the application for the original patent was lodged and the date on which the product was first authorised to be placed on the EU market, reduced by a period of five years. However, the duration of the SPC may not exceed five years from the date when it takes effect. Eventually, a pharmaceutical company that holds both a patent and an SPC can benefit from an overall maximum of 15 years of protection from the date its drug first obtains the authorisation to be placed on the EU market.
Figure 1: The EU’s incentives regime for the development and marketing of innovative pharmaceuticals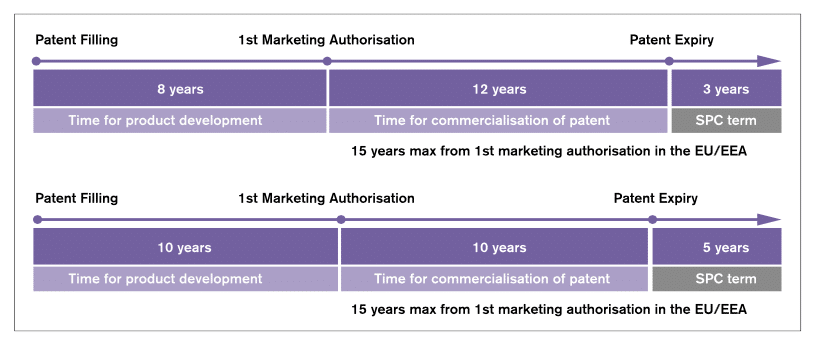
Source: own illustration derived from Pharmaceutical Compliance Monitor (2013). The figure shows two examples and how the SPC term is calculated based on the different times spent on development in two cases (in the first example 8 years and in the second example 10 years).
Should SPCs be Reformed?
The European Commission is now taking into consideration a change of the existing SPC legislation. It is following up on healthcare campaigners, the EU’s generic drugs industry and some EU health ministers who argue that pharmaceutical patents and other exclusivity protections ‘have gone too far, causing drug prices to skyrocket while patients struggle to access the latest treatments and cures’ (Colis 2017; Natsis 2017). While the generics industry wants to increase its sales, the motivation behind this initiative for governments is to reduce current or future expenditures on medicines.
Following the relevant communication of the European Commission (2017), the ambition is to (1) ‘create a level playing field for EU-based manufacturers of generic and biosimilar medicines by comparison with firms based in non-EU countries (both for export and for EU-market entry purposes).’ At the same time, the Commission aims at (2) ‘keeping a high level of SPC protection in the EU,’ which would, according to their view, ‘lessen reliance on imports of products that are essential to public health’ and help ‘innovative sectors to be globally more competitive and increase the attractiveness of Europe as a hub for innovation and manufacturing.’ Finally, the Commission (3) aims to ‘[m]aximise the benefits of the Unitary Patent system to those sectors relying on SPC protection, including a reduction of the fragmentation in the internal market that is associated with the current SPC system; and provide the upcoming Unified Patent Court with clear provisions reflecting best practice developed in Member States for these sectors.’
In addition to changes in existing SPC regulations, the Commission aims to ‘[m]aximise certainty and benefits of the Bolar patent exemptions, including a reduction of the fragmentation in the internal market that is associated with the current Bolar system’ and to ‘provide the upcoming Unified Patent Court with clear provisions reflecting best practice developed in Member States in relation to patent exemptions’ (European Commission 2017). The principle behind the Bolar exemption is that generic manufacturers should be legally permitted to take the necessary preparatory measures in order to be able to enter previously protected markets without delay once exclusive patent protection expires.
Where does the Industry Stand?
The generics industry emphasizes that a ‘manufacturing waiver for generic and biosimilar medicines during the SPC period would increase access to high quality medicines in unprotected markets, without changing the equilibrium between the originator and the generic and biosimilar medicines industries in the EU.’ It also claims that ‘an adapted legal environment that includes the SPC manufacturing waiver will boost investments in our sector and we stand ready to support job creation measures.’ (Medicines for Europe 2017a, 2017b) The generics industry also welcomes a study from early 2016 (released in October 2017) that was commissioned by the European Commission. This study concludes that the change in the EU’s current SPC regime will increase the net sales for the EU based pharmaceutical industry by 7.3 to 9.5 billion EUR and create 20,000 to 25,000 additional manufacturing jobs in Europe by 2025 (Medicines for Europe 2017c, CRA 2016).
R&D-intensive pharmaceutical manufacturers, most of which also produce generics, reject the idea of an SPC waiver. Industry representatives argue that it would weaken the EU’s incentives regime for pharmaceutical innovations and valuable new medicines. They also raise concerns about numerous practical challenges, specifically the inability of legal authorities to ensure that products manufactured under an SPC waiver are only exported. In addition, industry representatives are worried about products that are stockpiled for commercial purposes in the jurisdiction where manufacturing takes place, i.e. EU countries. Accordingly, an SPC exemption would effectively undermine the enforcement of IPRs, create additional administrative costs in the EU and increase litigation costs. Finally, the industry is worried about products that reach destinations other than the permitted countries, which would contribute to a further erosion of EU patent holders’ exclusive rights (IFPMA 2016).
Reviewing the Evidence – Studies of SPC Reform
As outlined by Pugatch et al. (2017), the case for an SPC waiver builds on the potential economic gains that have been outlined by Vicente and Simões (2014). These authors argue that an SPC manufacturing exemption would boost economic activity in the EU and create about 8,890 new direct jobs and 35,560 new indirect jobs. Vicente and Simões also state that, although the EU is one of the most IP-protected regions in the world for pharmaceuticals, high protection levels do not translate into greater rates of innovation.
A key argument brought forward by Vicente and Simões is that ‘the current European patent framework constitutes a serious obstacle to the competitiveness of local pharmaceutical manufacturers. Besides not being allowed to supply generic medicines to markets where patents have expired before expiration in Europe, the authors argue that these firms are also indirectly blocked from playing a relevant role in the supply of generic medicines to Europe. The authors also claim that EU-based pharmaceutical firms are ‘overtaken’ by competitors operating outside Europe since they ‘may launch their products immediately after SPC expiry.’ Vicente and Simões (2014, p. 46)
Vicente and Simões indeed make a strong point about national differences in IP protection, potential forum shopping and the exploitation of regulatory arbitrage. However, they do not provide any information about how much production is actually ‘overtaken’ by foreign suppliers and precisely which takeovers occur due to existing patent and SPC regulations.
Sussell et al. (2017) replicated the Vicente and Simões (2014) study. Contrary to the findings of Vicente and Simões, they estimate that potential job growth would be much smaller and, if uncertainty is taken into consideration, the estimated number for direct and indirect jobs is not statistically distinguishable from zero. Another study conducted by Logendra and Troein (2017) concludes that the actual market potential for EU generics companies to export to non-EU countries is relatively low as success on the markets depends to a large extend on local business practices, customer and patient preferences and is at the same time affected by prevailing trade barriers (for the high discriminatory impact of import tariffs on drugs, see Bauer 2017).
Logendra and Troein also argue that EU generics manufacturers face structural disadvantages as domestic companies often win the majority of business both initially and in the longer term, including the major non-EU markets such as Brazil, Russia and Turkey. Similar findings can be found in the European Commission’s pharmaceutical sector inquiry, which indicates that generics are slow in penetrating European markets. Differences in market shares between countries can, inter alia, be attributed to differences in local pricing and national reimbursement regulations, prescription policies, and diverse generic substitution laws (European Commission 2009), and they are to a large extent driven by well-considered product placement strategies of branded drug companies (Hess and Litalien 2005).
In addition, Logendra and Troein (2017) explain that generics manufactured in Europe are more likely to compete for market share with the original brands while at the same time EU’s branded drugs manufacturers would lose exports and market shares in non-EU countries. The authors also show that, for many drugs, SPC expiry dates in EU countries even precede those of non-EU countries. Finally, it is argued that the opportunities for European companies in relation to export markets are limited as, according to the authors, there is only a short or non-existent period of time to market these product that would be created by an SPC waiver.
The most comprehensive study on the impact of an SPC export waiver was only recently released by the European Commission (CRA 2016). It uses multiple methodologies and examines the impact of a SPC export waiver on both EU-based manufacturers of generics and producers of biosimilars. They also took into consideration business lost by branded manufacturers due to the entry of EU generics companies in non-EU markets. The authors estimate that additional sales of European branded and unbranded generics manufacturers would amount to 7.6 billion EUR by 2025 and 8.7 billion EU by 2030. For biosimilars, a different methodology was chosen. The estimated market size available to EU-based biosimilar producers – during the period between the protection expiry in the third country and the SPC expiry in Europe – ranges from 463 million EUR to 2.97 billion EUR by 2025. At the same time, innovative EU drug manufacturers are estimated to lose third country sales of up to 278 million EUR by 2025. Innovative European biologics manufacturers could lose sales of up to 1.7 billion EUR. In sum, CRA (2016) finds that the EU pharmaceutical industry could gain additional net exports of up to 8.56 billion EUR due to an SPC export waiver.
The authors note, however, that their estimates suffer from a serious lack of relevant trade, price and market share data for both branded drugs and generics medicines. While the methodology has generally been developed in a careful way, the numerical outcomes crucially depend on a number of simplifications, assumptions and approximations. For generics, two effects were taken into consideration: 1) a hypothetical market share that EU producers could achieve in eight major pharmaceutical markets (i.e. Canada, China, the US, Brazil, Russia, Japan, Turkey and Australia) based on the share of EEA imports to each of these countries and these countries overall pharmaceutical sales, and 2) additional sales due to market share effects that result from first-mover advantages, which, importantly, tend to generate relatively high numerical effects on the study’s ‘lost sales’ estimates.
It is estimated that generics companies entering a market one year after the first generic entrant would be able to reach a market share of up to 27 per cent of that of the first entrant 12 months after the entry of the later entrant, and up to 36 per cent 24 months after the entry of the later entrant. Market shares are expected to decline considerably the longer it takes for foreign market entrants to enter the market. The authors acknowledge that ‘there could be factors other than the delay related to the SPC term that affect the time to entry into an export market, such as e.g. delays in preparing the dossier and receiving regulatory approvals, delays associated with pricing and reimbursement to the extent that these apply in third countries, among others.’ (CRA 2016, p. 111). These factors could impact on the time it takes for third market entry, but would not be affected by the date of protection expiry in third countries. Defence strategies of incumbent companies, including originators companies who might engage in ‘flanking’ or ‘value-extending’ strategies (as outlined by Glazier et al. 2016), are not taken into consideration.
Concerning the calculation of market shares, the authors acknowledge that there is ‘no reliable data on the share that European generic and biosimilars achieved in sales in third countries,’ which is why calculations in their study are merely based on aggregate EEA export data that do not distinguish between generics drugs, biosimilar drugs and branded drugs. Accordingly, it is assumed that the ratio of imported generics/biosimilars to originator products was the same as the ratio of generic pharmaceutical sales to originator sales in the importing country (CRA 2016, p. 113).
In other words, the estimations provided do not account for dynamic changes in market shares. Such changes would emerge over time due to altering forces of supply and demand. For example, there would be price and volume changes due to the displacement of branded drugs imported from the EU. What is important from the perspective of EU governments: price and volume changes for branded drugs would occur within the EU due to a strategic reconsideration of prices set by pharmaceutical companies in countries where their products are still protected by market exclusivity rights. Moreover, since the data applied does not allow for a differentiation of branded and unbranded generics, the conclusions do not differentiate between ‘pure’ generics manufacturers and brand-name manufacturers that also serve generics markets (CRA 2016, p. 112).
For biosimilars, the authors also highlight that they had to work on the basis of simplifications, assumptions and approximations due to the lack of sufficient market data for EU and third countries. EU market penetration rates, for example, have been applied for third countries as third country penetration rates are not available for biosimilars. A similar lack of data is reported for the actual volume of sales of biosimilars in third countries.
Non-availability of data is also reported for the volume of branded drugs and branded biosimilars that EU exporters sell in third countries. Similarly, the authors were unable to collect data for prices and price elasticities in third countries, which is why they assume constant prices for originator products after the entry of generics due to the expiration of the SPC protection. At the same time, it is argued that ‘recent papers have found that branded medicines prices do drop somewhat or remain flat following protection expiry in the US, Canada and European markets’ (CRA 2016, p. 135), indicating that originator manufacturers prices’ are actually likely to be exposed to downward pressure due to rising generics competition.
To sum it up, even though this and other studies are carefully done, the results should be interpreted with caution. Reliable quantitative analysis requires reliable data. Reflecting the lack of data and methodological choices to circumvent the absence of hard quantitative facts, the results of all available studies differ substantially in size and the direct and indirect effects on EU production and employment. In addition, the analyses suffer from a lack of sufficient industry data, particularly projections and case studies from individual EU generics manufacturers and impact analyses provided by their industry bodies credibly backing the generic industry’s promise to invest and expand exports.
What unites available studies, however, is that they all point to marginal effects on overall EU economic activity if a waiver is introduced. At the same time, they also ignore the dynamic effects of changes in the EU’s incentive regime for pharmaceutical companies that engage in knowledge-intensive research and development activities, i.e. the effects on prices of drugs that still benefit from patent protection and exclusivity rights. Importantly, all these studies fail to address the opportunity costs that an SPC export waiver would create for patients and public health systems of EU Member States as the result of strategic changes in product placement, pricing and investment behaviour by innovators.
The Opportunity Costs of an SPC Export Waiver
There are opportunity costs that would arise from the implementation of an SPC export waiver. In this Section, we will discuss the major determinants of the ‘economic value of a patent’ and companies’ incentives to invest in the research and development of new drugs. We argue that the EU’s innovative pharmaceutical manufacturers, irrespective of whether they develop and manufacture originator drugs or innovative biosimilars, are very likely to adapt their business models in a way that either contains costs and/or research and development activities or by attempts to increase revenues by increasing prices.
We also argue that it is highly unlikely that the EU’s innovative pharmaceutical manufacturers merely scale-down production in the EU due to lower exports to third countries. Generally, if the effective exclusivity period is shortened, the manufacturers of branded drugs will have to raise revenues over a shorter period of time. Hence, they are likely – and have proven to do so in the past – to raise prices for some of their products. Accordingly, the ‘unintended’ and so far ‘unattended’ consequences of the waiver would go far beyond the redistribution of sales between branded and generics drugs, which seems to be an intention of some who argue in favour of a waiver.
Like any other change in regulation, companies will respond to an SPC waiver by making changes in their marketing, pricing and product placement strategies. Some may reduce investments in R&D, but most companies – presented with a shorter exclusivity period – will respond by increasing the prices during the period in which they still enjoy market exclusivity rights. By comparison, in a mere scale-down scenario (as implicitly assumed by the authors of existing studies), the EU’s innovative drug and biosimilar manufacturers would not change their product placement behaviour to exploit alternative sources of revenues. In other words, they assume that lower revenues and lower profits will prompt no change in business strategy. This, however, is highly unrealistic. The two chief opportunity costs that policymakers need to consider are:
- Higher prices paid by EU customers (public healthcare systems) for several medicine products and/or
- Lower levels of investment in drug development in primarily high-cost EU Member States
Declining Commercial Value of Patents
It is expensive and time consuming to develop a new drug. The rate of failure in drug development is extremely high as researchers have to study thousands of compounds before turning substances into a marketable product that generates value for patients. For clinical drug development success rates, for example, BIO Industry Analysis (2016) calculates that the overall likelihood of approval (LOA) from the first clinical trial phase (Phase I) was only 9.6 per cent between 2006 and 2015, and 11.9 per cent for all indications outside the field of oncology. The report also states that Phase II clinical programs continue to suffer from the lowest success rate of the four development phases, with only 30.7 per cent of developmental candidates advancing to Phase III.
The effective commercial value of a pharmaceutical patent is driven by two factors: costs and revenues. Costs are composed by R&D costs, product development costs, international drug approval costs, the costs of manufacturing, and costs related to marketing and the distribution of the drug. Revenues, on the other hand, are primarily determined by product life cycles, time of marketing exclusivity, prices and, usually, national market size(s). In general, the profitability of a certain drug depends on the capability of patent owners to raise sufficient revenues over a given period of time, i.e. revenues that exceed the fixed drug development and variable production and marketing costs.
In practice, the economic success of a new drug depends on a number of uncertainties that affect the cost structure of companies and their effective ability to collect sufficient revenues in fragmented national markets over a certain time period. After all, these are the factors that impact on a company’s (or investors’) judgement about whether selling a marketable drug will eventually allow the company to successfully recoup its research and market development expenditures – and these considerations feedback on the managerial decisions underlying various research projects for new drugs. Although many features of pharmaceutical markets are far from transparent for ‘outsiders’, various data and several studies indicate that several important market fundamentals significantly deteriorated from the perspective of innovative pharmaceutical manufacturers in the past decade. In the following, we will consider:
a) Trends in drug development costs, and
b) Trends in companies’ price-setting practices and national reimbursement policies
a) Longer Times and Higher Costs: Trends in Drug Development Costs
Many studies indicate that the cost and time of drug development increased tremendously since the 1960s. Even though there have been many advances in pharmaceutical research over the past 60 to 70 years, efficiencies in commercial drug development research declined dramatically. Scannell et al. (2012), for instance, report that the number of new drugs approved per billion USD spent on R&D has ‘halved about every 9 years’ since the 1950s. This decline corresponds to an 80-fold productivity drop after adjustment for inflation.
Investigating the underlying forces of the ‘loss of productivity’ in drug research, Scannell et al. discuss four factors that they consider to be primary causes: 1) the ‘better than the Beatles’ problem, according to which shifts in R&D to new therapeutic fields with lower approval probabilities cause higher attrition rates. This is to a large extent driven by incentives in reimbursement systems and health technology assessments (HTAs) that are intended to trigger developers to go for the ‘difficult’ parts, especially oncology and ‘orphan’ drugs, and to pay on the basis of outcomes (Adkins et al. 2017). These are exactly the therapeutic areas where it is difficult to pass the market approval tests, with incentives of which policymakers do not at all know whether they are going to contribute to the dual goals of improving outcomes and controlling costs. 2) the ‘cautious regulator’ problem, which arises due to regulators increased risk awareness (lower risk tolerance) over time and, therefore, the higher trial and patient safety assurance costs (for additional regulatory provisions, see also EU 2012 and WHO 2012). 3) a ‘throw money at it’ tendency, which means that companies assign many scientific resources to certain projects in order to be the first to launch a new drug. 4) the ‘basic research–brute force’ bias, according to which there is a tendency in the industry to overestimate the ability of advances in basic research and brute force screening methods.
The authors state that these factors largely contributed to the extensive decline in pharmaceutical research productivity. In addition, the authors state that lower performance rates in general research can be attributed to the industry’s trend of seeking ‘precise effects’ from molecules rather than broader therapeutic potential, whereby the targeting of certain therapeutic effects comes with longer and more complex trial and error activities. These issues have not been taken into consideration by those who provided analytical work on the effect on an SPC export waiver.
DiMasi et al. (2016) argue that higher company out-of-pocket costs for individual drugs and higher failure rates for drugs tested in human subjects were the major reasons behind rising drug development costs. Higher out-of-pocket expenditures arose from higher clinical trial complexity, larger clinical trial sizes, higher cost of inputs from the medical sector used for development, changes in protocol design to include efforts to gather health technology assessment (HTA) information, and testing on comparator drugs to accommodate payer demands for comparative effectiveness data. The authors calculate an increase in drug development costs of 145 per cent from 2003 to 2013. Taken together, these developments heavily impacted on the drug development costs of those pharmaceutical companies that strongly engage in scientific research in innovative medicines.
All the above-mentioned effects impact on different stages of development and have substantially inflated the average cost of drug development over time. Available empirical evidence suggests that pharmaceutical research and development cost skyrocketed in the past 20 years. Several recent studies indicate that the costs of drug development often amount to up to several billions of USD (EUR) per drug (view Table 1 for an overview of various ‘average cost’ estimates).
Mestre-Ferrandiz et al. (2012), for example, report that the costs of pharmaceutical R&D activities vary for various kinds of drugs, depending on therapeutic area, firm sizes and whether the molecule is a ‘traditional’ chemical compound or a biologic. On average, the authors find that drug development costs amount to an average 1.5 billion USD expressed in 2011 prices. Herper (2013) presents R&D cost data for 98 companies and (roughly) estimates drug development costs to amount to up to 5.3 billion USD per approved drug. The study by DiMasi et al. (2016) estimates the pre-tax capitalized R&D cost per approval of up to 2.6 billion USD. When post-approval R&D costs are included, the average cost rise to 2.9 billion USD. The estimates reflect the full cost of gaining regulatory marketing approval for a new drug (see Table 1).
For generics manufacturers, the research and development cycle is much less complex, shorter and cheaper relative to the branded R&D cycle (for an illustration see, e.g., MUFG 2016). Compared to innovative brand-name drugs, the risk of failures in the R&D process for generic drugs is low as the safety and efficacy of the active ingredient has already been established. Accordingly, the relative simplicity of generics companies’ R&D processes is a major driver of and integral to the low-cost nature of generic drugs.
In addition, contrary to branded drug manufacturers, generics companies tend to show a flat development of R&D expenditures due to their motivation to develop ‘marketable formulations of a large number of drugs that are either out of patent or may be soon’ out of patent (Basu et al. 2008, p. 34). Similarly, generics companies tend to show lower expenditures for marketing and the distribution of drugs than both innovative brand-name drug manufacturers and biotech drug manufacturers.
Table 1: Studies providing estimates for the R&D costs of new drugs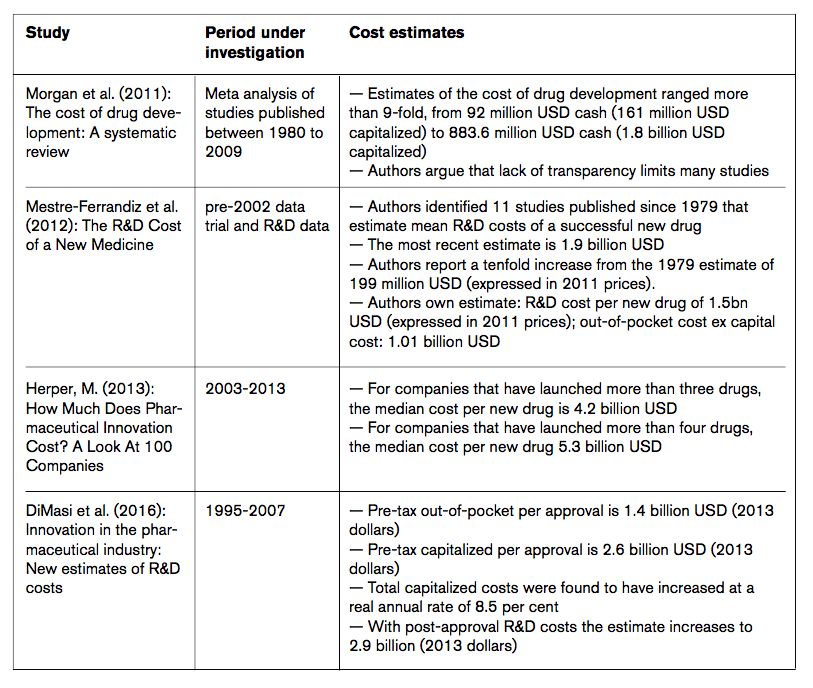
Source: ECIPE research.
Although the economic returns realised by pharmaceutical firms are a frequent topic of debate, industry data only rarely inform stakeholders about past and recent developments of relevant indicators such as reimbursement rates, prices, sales figures and the present commercial value of medicinal products (for a general discussion see, e.g., Hannah and Phillips 2017). Similarly, although a large body of research has highlighted the riskiness of investments in new drugs, only little attention has been paid to what happens once drugs are developed, i.e. the commercialisation phase of the drug. Industry data suggest, however, that ‘big pharma’ is far from being as profitable now as they were in the past. These data indicate that innovative pharmaceutical companies are not generating profits of a kind that suggest they are just capturing rents from patents.
In an attempt to shed light on the path of economic returns of various prescription medicines, Berndt et al. (2014) studied the economic returns of four cohorts of new prescription drugs that were launched in the US between 1991 and 2009. The authors took into consideration variations in revenues and fluctuations in average research and development (R&D) and other costs to determine patterns in the rewards for investment in pharmaceutical innovations. Their results indicate that ‘average present values of lifetime net economic returns were positive and reached a peak with the 1995–99 and 2000–04 new drug cohorts.’ They also show that the returns on innovation have ‘fallen sharply since then, with those for the 2005–09 cohort being very slightly negative and, on average, failing to recoup research and development and other costs.’ The riskiness of investment is also reflected by the volatility of revenue streams, which are, generally, far more volatile than the costs of R&D. Raising concerns that pharmaceutical companies could not be able to operate under rising returns in the future, the authors argue that pharmaceutical manufacturers may not maintain the rates of investments needed to sustain biomedical innovation.
In addition to these general trends, competitive pressure from generics companies increasingly impacts on innovative manufacturers’ revenues and profitability rates, i.e. their capacity to invest in new research. Generics companies entering new markets exert downward pressure on prices and tend to erode incumbents’ market shares. According to an industry analysis of MUFG (2016), the average share of branded drugs eroded to only 16 per cent after 12 months post generic entry for the period 2011 to 2012, compared to a relatively high 51 per cent in the late 1990s. As a general rule, MUFG states that generics competition tends to reduce pricing/market share of a branded drug by more than 80 per cent after the first 12 to 18 months from the time of market entry of a generic product.
MUFG (2016, p. 10) also argue that ‘generic manufacturers have been becoming increasingly aggressive and have successfully shortened market exclusivity periods.’ The authors report an increasing number of filings through which generics manufacturers seek to invalidate branded patents. These filings became much more frequent and now generally happen much earlier than in the late 1990s and early 2000s, effectively contributing to the erosion of branded drug’s market exclusivity periods.
Generics competition also impacts on those products that are still protected by patents in cases where generic substitution is partially possible. As generics competition increased significantly in the past decade, the effective commercial value of a patent decreased. As a consequence, there is additional pressure on innovative pharmaceutical companies to cut research and production costs or to increase prices of products for which competition is less intense.
Cost estimates insofar matter as they are a major issue in the international debate about the appropriateness of pharmaceutical prices and the magnitude of regulations of intellectual property rights for pharmaceutical products. It should be noted, however, that research in this field generally suffers from a lack of firm-level data, project and product resource assignments, but also information about the costs of approval procedures imposed by regulatory bodies.
Additionally, there is broad consensus among scholars, regulators and industry experts that the time needed to obtain necessary market approvals for an innovative product increased substantially over time, and that these procedures eat time for the effective duration of the underlying patent. Notably, the regulatory requirements are unlikely to be relaxed in the future. As outlined by EMA and national regulatory experts, ‘[r]egulators should not, for the sake of affordability, yield to pressure to lower standards. But it’s also inappropriate for them to be oblivious to the growing budget pains caused by newly authorized products (Eichler et al. 2016, p. 1808). Consequently, regulatory costs are highly unlikely to drop in the future, leaving innovative pharmaceutical companies with high out-of-pocket expenditures, and a systematic healthcare policy-induced disadvantage vis-à-vis generics manufacturers.
b) Lower Revenues: Price-setting Behaviour and Trends in National Reimbursement Policies
The global pharmaceutical industry is highly regulated by governments focusing on both safety and costs. Governments have become less willing to reimburse the full market costs of a new drug. Increasing public budget deficits and higher government debt burdens triggered an increased focus on reducing healthcare spending and pricing fuelled by aging and growing populations, the proliferation of chronic diseases and a stronger focus on care quality and value (Abbott 2016; Deloitte 2016; OECD 2015, Remuzat et al. 2015).
While globalisation has increased the number of potential customers around the world, most markets that have been rapidly growing do not have market-approval and reimbursement systems that lead to higher sales for drug companies corresponding with the rise of affluence in those countries. Globally, governments are increasingly pushing for value-based payment and reimbursement systems. At the same time, most countries are instituting various types of drug price controls and continue to look for other ways to contain public spending on medicines, e.g. reference pricing, fee-based systems, centralised bidding-systems and quotas for the use of generics (Deloitte 2016).
Research-intensive and innovative pharmaceutical companies face the challenge that their main customer in most countries are governments that do want to lower their expenditures. Most public healthcare systems, including those in the EU, now enforce several cost containment policies aiming to decrease average prices paid. As a result, the effective value of protection has gone down because of monopsonic markets[1] that are strongly guided by the reduction of expenditures.
For the EU market, lawmakers tried to harmonise regulatory processes. The European Medicines Agency is now responsible for granting marketing authorisation. However, even though there is a ‘recognised need to move toward a more objective and collaborative reimbursement environment for new medicines in Europe,’ the 28 EU member states still have national decision-making power over drug prices paid by the government and national reimbursement practices (Allen et al. 2017, p. 384). Thereby the lack of centralisation is reflected by an overly complex patchwork of national regulations and guidelines, which are at the root of significant differences in prices for medicines among EU countries.
As outlined by O’Donnell (2015), no two countries regulate their pharmaceutical markets it the same way, allocating their national healthcare budgets and follow their national healthcare policy priorities. The authors also highlight that most governments indeed ‘officially’ aim for more comparable prices, but that at the same time the ‘information they have is often insufficient, and the methods they use for making their comparisons vary dramatically.’ For outsiders, it is generally impossible to trace prices and price development.
Using publicly available data, Allen et al. (2017) study nine European reimbursement systems by assessing the relationship between the regulatory and reimbursement decision-making organisations (the pricing authorities). Their findings show that pharmaceutical manufacturers are still required to submit multiple applications to individual EU Member State for the evaluation of ‘added value’ and reimbursement assessments, which can result in varying reimbursement recommendations.
These processes and the time it takes to negotiate with many national agencies add to the shortening of the effective exclusivity period, with an impact on prices charged. Pinyol et al. (2015), for example, study the duration of the reimbursement process in Spain. They find that the time from EMA approval to reimbursement increased significantly from 230 days in 2009 to 431 days in 2013. Similarly, for the period 2009 to 2013, Mycka (2014) find that the time from regulatory approval to the first launch of a drug in the US averaged 39 days. For EU countries (Germany, France, the UK, Italy and Spain), Germany was fastest while Italy was slowest (16 versus 66 weeks).
As a consequence, aware of the regulatory heterogeneity and an ever-changing patchwork of policies and regulations, manufacturers ‘often submit applications first to markets where they are likely to achieve a higher price.’ (Allen et al. 2017, p. 384) In other words, as indicated by Morton and Kyle (2012), pharmaceutical manufacturers tend to negotiate over prices and launch new products in high-price countries first, allowing them to positively affect any reference price used by later countries.’ Allen et al. (2017) therefore recommend a more harmonized HTA environment within the EU. This, however, remains far from the reality today.
The high degree of heterogeneity in EU reimbursement regulation is reflected by the high degree of price variation in the EU. The high degree of price variation shows that pharmaceutical manufacturers of branded and generics drugs are still able to negotiate set prices on a relatively discretionary basis. Zaprutko et al. (2017), for example, show that 2012 drug prices varied significantly between EU Member States, whereby the highest prices were found for Germany. At the same time, comparatively low drug prices were found for various countries – even though the cheapest products were not found in the Member States with lowest per capita incomes, i.e. Bulgaria or Romania. Wouters et al. (2017) find that prices and market shares of generics vary widely across Europe. Prices charged by manufacturers in Switzerland are, on average, more than 2.5 times those in Germany and more than 6 times those in the UK. The share of generics prescriptions ranges from 17 per cent in Switzerland to 83 per cent in the UK.
Rémuzat et al. (2015) conducted an analysis of EU Member States’ External Reference Pricing (ERP) systems, which are widely applied either for all marketed drugs or for specific categories of medicines eligible for reimbursement. They find a great variation in the calculation methods used to compute the prices for reimbursement (15 countries used the average price, 7 countries used the lowest price, and 7 countries used other calculation methods). The authors further report a lack of reliable sources of price information because of price heterogeneity, exchange rate volatility and discounts, which impact on the pricing strategies from pharmaceutical companies.
Importantly, Rémuzat et al. state that the reference pricing system has become an incentive for pharmaceutical companies to ‘adopt international pricing strategies. Launch sequence strategies are used to delay or avoid launching new drugs in countries with potential lower prices, especially if they are small markets referenced by countries with larger markets.’ (Rémuzat et al. 2015, p. 9)
The Opportunity Costs of an Export Waiver
Policy reforms always come with opportunity costs – and it is remarkable that those have not received more attention in the debate concerning the SPC export waiver. If such a reform would happen, opportunity costs would emerge for EU Member States due to changes in innovative pharmaceutical manufacturers’ product placement, pricing, production and investment strategies.
Even though the precise trade diversion impact of an SPC export exception is hard to predict due to a profound lack of industry data, an SPC waiver would undeniably impact on pharmaceutical manufacturers’ opportunity-risk assessments regarding the future marketability of drugs in EU markets and third countries. A relaxation of the EU’s existing SPC regime generally comes with an increased risk of lower revenues in non-EU countries and, ultimately, a higher risk of lower revenues on EU markets, as in some cases generics companies would be able to penetrate EU markets more readily after the expiration of EU market exclusivity of protected branded drugs (due to stockpiling).
Like other ‘nimble’ industries, the pharmaceutical industry responds to changing circumstances. Past experiences undeniably show that the industry pursued various strategies to cope with regulatory heterogeneity within the EU and between the EU and third countries. Therefore, an SPC export waiver would most likely cause the EU’s innovative manufacturers to reconsider their global R&D and manufacturing activities as well as their global and, primarily, EU product placement and pricing strategies to compensate for (the risk of) lower revenues abroad.
Measures to address the changing circumstances following an export waiver would include:
- Divest-EU (cost containment strategy): EU pharmaceutical manufacturers scale down their overall R&D activities in high-cost EU countries to the benefit of low-cost countries inside the EU.
- Divest-non-EU (cost containment strategy): EU pharmaceutical manufacturers outsource both R&D as well as manufacturing capacities to low-cost countries outside the EU.
- Raise-prices-EU (revenue stabilisation strategy: EU pharmaceutical manufacturers increase the prices in those markets in which their products still enjoy market exclusivity rights. In the case of lost sales of certain products due to an EU SPC export waiver, this would be the EU market, primarily high-income (Western and Northern) EU Member States.
The reconsideration of research and production capacities would primarily affect those countries where large and mid-size life science companies are currently heavily invested, i.e. primarily high-cost Western Europe (KMPG 2016).
An SPC export waiver would come with the risk of higher pharmaceutical prices in EU Member States where market exclusivity is still active, without creating new incentives for post-patent competition. This is primarily true for high-income Member States, but also for ‘referencing’ low-income Member States.
Adaptations in innovative manufacturers’ product pricing, negotiation and launch sequence strategies are most likely to affect the EU’s most affluent, most populous Member States. In these countries, patients and governments are likely to face higher prices of branded drugs and branded generics. In other words, the EU governments of (primarily) Western European countries are unlikely to see any savings on their drug bill because of an SPC waiver.
[1] A monopsony is a market structure in which only one buyer interacts with many would-be sellers of a particular product.
Concluding Remarks
Despite good efforts, little of value can be said about the aggregate effects on European exports and value-added from an SPC waiver. The fact is that many generics producers already produce in other countries and are unlikely to relocate much production from low-cost countries to the EU. The generics industry’s promise to invest in the EU to be able to export more from the EU are vague and have not been backed by industry data, let alone credible commitments at firm level.
An export waiver is unlikely to help the EU’s generic producers to move from their current model, which is subject to squeezed margins, to new models in higher-margin niches. The industry is faced with other structural problems that have reduced their productivity and competitiveness, both in Europe and elsewhere: declining prices, increased competition, and a dwindling number of blockbuster pharmaceuticals going generic (Bujnoski 2017).
What seems obvious, however, is that the economic effect of an SPC export exemption is primarily to redistribute value-added from upstream innovators to downstream producers within the EU: the generics industry would increase exports and generate more value-added while those producing drugs under an SPC would have deteriorating sales and generate less value added in Europe. Furthermore, important dynamic effects would arise from what also seems to be an obvious observation: the producers of drugs with SPC exclusivity would have to recoup their investments and generate profits in a shorter period of time, which naturally would involve attempts to raise these products’ prices.
In the past decades, several factors already contributed to a significant erosion of the effective market exclusivity period of patented drugs. SPCs were intended to compensate for longer market-approval processes. If that period is shortened, the erosion of market exclusivity will be reinforced. For innovative originators, it is therefore difficult to find other means for doing so than raising the prices of their products.
The EU’s reference pricing system has already become an incentive for pharmaceutical companies to adopt launch sequence strategies that delay or avoid launching new drugs in countries with potential lower prices, especially if they are small markets referenced by countries with larger markets. Accordingly, given what we know from strategies to overcome regulatory fragmentation and market sequencing strategies in the EU, the most promising way to achieve this is to get the EU’s high-income countries to pay more. Therefore, an SPC export waiver is likely to cause higher drug prices for governments and patients in countries such as Germany, the UK, France, the Nordics, and the Benelux.
The European Commission’s current approach to reform the existing SPC regime stands in opposition to the objectives of the EU’s current trade and investment policy focus for intellectual property rights. The signalling effect of an SPC export exemption could evoke policies in third countries that stand in opposition to EU trade policy interests regarding the protection of intellectual property rights in general. It would not contribute to a convergence of IP protection regimes to bridge global gaps in IP protection and to avoid regulatory forum shopping.
References
Abbott, A. (2016), Legal Threats to Strong Returns on Pharmaceutical Patents Grow, Threatening Innovation, IPWatchdog article from 17 January 2016, available at http://www.ipwatchdog.com/2016/01/17/legal-threats-to-strong-returns-on-pharmaceutical-patents-grow/id=65013/, accessed on 10 November 2016.
Adkins, E. M., Nicholson, L., Floyd, D., Ratcliffe, M. and Chevrou-Severac, H. (2017), Oncology drugs for orphan indications: how are HTA processes evolving for this specific drug category?, ClinicoEconomics and Outcomes Research, 9, p. 327–342.
Allen, N., Liberti, L., Walker, S. R. and Salek, S. (2017), A Comparison of Reimbursement Recommendations by European HTA Agencies: Is There Opportunity for Further Alignment?, Frontiers in Pharmacology.
Basu, P., Joglekar, G., Rai, S., Suresh, P. and Vernon, J. (2008), Analysis of Manufacturing Costs in Pharmaceutical Companies, Journal of Pharmaceutical Innovation, Volume 3, 2008.
Bauer, M. (2017), The Compounding Effect of Tariffs on Medicines: Estimating the Real Cost of Emerging Markets’ Protectionism, ECIPE Policy Brief 1/2017.
Berndt, E. R., Nass, D., Kleinrock, M. and Aitken, M. (2014), Decline In Economic Returns From New Drugs Raises Questions About Sustaining Innovations, Health Affairs.
Bio Industry Analysis (2016), Clinical Development Success Rates 2006-2015, available at https://www.bio.org/sites/default/files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf.
Bujnoski, J. (2017), Can Biosimilars Increase The Profitability Of Generics Manufacturers?, published on 7 September 2017, available at https://www.biosimilardevelopment.com/doc/can-biosimilars-increase-the-profitability-of-generics-manufacturers-0001, accessed on 20 November 2017.
CHAFEA (2017), Euripid Project “Statistical data for medicinal product pricing” reaches half-time, available at http://ec.europa.eu/chafea/news/news492.html, accessed on 10 November 2017.
CRA (2016), Assessing the economic impacts of changing exemption provisions during patent and SPC protection in Europe, study commissioned by the European Commission, prepared by Charles River Associates.
Colis, H. (2017), Drug lobby’s market protections, available at: https://www.politico.eu/article/future-of-pharma-incentives-fine-line-between-incentives-and-favoritism-drug-research/, accessed on 20 November 2017.
Eichler, H. G., Hurts, H., Broich, K. and Rasi, G. (2016), Drug Regulation and Pricing — Can Regulators Influence Affordability? The New England Journal of Medicine.
EMA (2012), Background to the pharmacovigilance legislation, available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000491.jsp, accessed on 10 November 2017.
European Commission (2017), Inception Impact Assessment on ‘Optimising the Internal Market’s industrial property legal framework relating to supplementary protection certificates (SPC) and patent research exemptions for sectors whose products are subject to regulated market authorisations’, 15 February 2017, DG Growth.
European Commission (2009), Pharmaceutical Sector Inquiry—Final Re- port. Brussels, Belgium: European Commission.
European Parliament (2016), EU Options for Improving Access to Medicines, Study for the ENVI Committee, 14 July 2016.
European Parliament (2011), Differences in Costs of and Access to Pharmaceutical Products in the EU, Directorate General for Internal Market Policies, Policy Department A, Economic and Scientific Policy.
Glazier, F., Fezza, T. and Reynolds, J. (2016), Loss of Exclusivity: Strategies to Maximize Product Value, article published on PharamaExec.com, 9 November 2016, available at: http://www.pharmexec.com/loss-exclusivity-strategies-maximize-product-value, accessed on 20 November 2017.
Deloitte (2016), 2016 Global health care outlook – Battling costs while improving care.
DiMasi, J. A., Grabowski, H.G and Hansen, R.W. (2016), Innovation in the pharmaceutical industry: New estimates of R&D costs, Journal of Health Economics (47), May 2016, p. 20-33.
IFPMA (2016), Policy Position concerning the ‘Export Manufacturing Exemption During SPC/PTR Extended Patent Term: Global Position, 12 September 2016.
Hannah, L. and Phillips, J. (2017), Is the Current UK System of Pharmaceutical Price Regulation Working?, Hausfeld.
Herper, M. (2013), How Much Does Pharmaceutical Innovation Cost? A Look At 100 Companies, Forbes – Pharma and Healthcare, 11 August 2013, available at: https://www.forbes.com/sites/matthewherper/2013/08/11/the-cost-of-inventing-a-new-drug-98-companies-ranked/#52dfb4aa2f08, acceessed on 14 November 2017.
Hess, J. and Litalien, S. (2005), Battle for the market: Branded drug companies’ secret weapons generic drug makers must know, Journal of Generic Medicines, 1, p. 20-29.
Kashyap, U. Ni., Gupta, V. and Raghunandan, H. V. (2013), Comparison of Drug Approval Process in United States & Europe, Journal of Pharmaceutical Sciences and Research, Volume 5, 2013.
KPMG (2016), Site Selection for Life Sciences Companies in Europe, 2016 Report.
Logendra, R. and Troein, P. (2017), Assessing the impact of proposals for a Supplementary Protection Certificate (SPC) Manufacturing Exemption in the EU, White Paper published by Quintiles IMS.
Mestre-Ferrandiz, J., Sussex, J. and Towse, A. (2012), The R&D Cost of a New Medicine, Office of Health Economics, December 2012.
Medicines for Europe (2017a), European Parliament Recognises the Major Role of Generic, Biosimilar and Value-added Medicines in Improving Access, Brussels, March 2017.
Medicines for Europe (2017b), Stimulating Investment in European Generic, Biosimilar and Value-added medicines through a transparent legal environment, London, 22 March 2017.
Medicines for Europe (2017c), Commission Sponsored Study Shows Supplementary Protection Certificate (SPC) Manufacturing Waiver will create Jobs, Strengthen the Pharmaceutical Sector and Reduce Medicine Costs.
Medicines for Europe (2017d), Position Paper External Reference Pricing for Generic and Biosimilar Medicines, 1 July 2017.
Morton, F. S., and Kyle, M. (2012), Markets for pharmaceutical products, Health Economics, 804, 2012.
MUFG (2016), Global Generic Pharmaceutical Industry Review, Bank of Tokyo-Mitsubishi UFJ, February 2016.
Mycka, J., Dellamano, R., Lobb, W., Dalal, N., Pereira, E., Dellamano, L. and Sagaydachnaya, O. (2014), Regulatory Approval to Patient Access, an Evaluation of EU5 and us National Timing Differences, Value in Health, 17, p. A794–A795.
Natsis, Y. (2017), New game, new rules: Pathways for better and affordable medicines in Europe, available at https://epha.org/new-game-new-rules-pathways-for-better-and-affordable-medicines-in-europe/, accessed on 20 November 2017.
O’Donnell, P. (2015), The Great Euroepean Drug Problem, article published by Politico, available at: https://www.politico.eu/article/the-great-european-drug-problem-high-priced-products-expenditure/, accessed on 10 November 2017.
OECD (2015), Fiscal Sustainability of Health Systems – Bridging Health and Finance Perspectives, OECD Publishing, Paris.
Pharmaceutical Compliance Monitor (2013), Exclusivity from Pediatric Data – EU Overview, available at http://www.pharmacompliancemonitor.com/exclusivity-from-pediatric-data-eu-overview/6036/, accessed on 5 October 2017.
Pinyol, C., Valmaseda, A., Gomez-Ulloa, D. and Restovic, G. (2015), Duration of the Reimbursement Process in Spain for Innovative Drugs Approved by the European Medicines Agency during the Period 2008-2013, Revista Espanola Salud Pública, 89, p. 189-200.
Pugatch, M., Torstensson, D. and Laufer, M. (2017), Unintended Consequences: How introducing a manufacturing and export exemption to supplementary protection certificates would weaken global standards of IP protection and result in direct losses to Europe’s research-based biopharmaceutical industry.
Putzeist, M. (2013), Marketing Authorization of New Medicines in the EU – towards evidence-based approvement, Thesis Utrecht University -with ref.- with summary in Dutch. The studies presented in this thesis were performed in the context of the Escherproject (T6-202), a project of the Dutch Top Institute Pharma. Parts of this thesis were performed in collaboration with the Dutch Medicines Evaluation Board, Utrecht, The Netherlands Julius Center for Health Sciences and Primary Care, Utrecht, The Netherlands European Medicines Agency, London, United Kingdom.
Rémuzat, C., Urbinati, D., Mzoughi, O., El Hammi, E., Belgaied, W. and Toumi, M. (2015), Overview of external reference pricing systems in Europe, Journal of Market Access and Helath Policy.
Scanell, J. W., Blanckley, A., Boldon, H. and Warrington, B. (2012), Diagnosing the Decline in Pharmaceutical R&D Efficiency, Nature Reviews, Drug Discovery, Volume 11, March 2012.
Sheridan, D. and Attridge, J. (2013), The Impact of Therapeutic Reference Pricing on Innovation in Cardiovascular Medicine, PharmacoEconomics, Volume 24, 2013.
Sussell, J. A., Tebeka, M. G., Jena, A. B. and Vanderpuye-Orgle, J. (2017), Reconsidering the economic impact of the EU manufacturing and export provisions, Journal of Generic Medicines, Vol 13(2), p. 1-17.
Vicente, V. and Simões, S. (2014), Manufacturing and export provisions: Impact on the competitiveness of European pharmaceutical manufacturers and on the creation of jobs in Europe, Journal of Generic Medicines, Vol. 11(1–2), p. 35-47.
WHO (2010), Minimum Requirements for a functional Pharmacovigilance System, World Health Organization, available at http://www.who.int/medicines/areas/quality_safety/safety_efficacy/PV_Minimum_Requirements_2010_2.pdf, accessed on 10 November 2017.
WIPO (2016), World Intellectual Property Indicators 2016, Economic and Statistics Series of the World Intellectual Property Organization.
WTO (2000), DS114: Canada – Patent Protection of Pharmaceutical Products.
Wouters, O. J., Kanavos, P. G., McKee, M. (2017), Comparing Generic Drug Markets in Europe and the United States: Prices, Volumes, and Spending, The Milbank Quarterly, Vol. 95, No. 3, 2017.
Zaprutko, T., Kopciuch, D., Kus, K., Merks, P., Nowicka, M., Augustyniak, I. and Nowakowska, E. (2017), Affordability of medicines in the European Union, PLOS One, open access article.